- Hemoglobin
- The oxygen-carrying pigment and predominant protein in the red blood cells. Hemoglobin forms an unstable, reversible bond with oxygen. In its oxygenated state it is called oxyhemoglobin and is bright red. In the reduced state it is called deoxyhemoglobin and is purple-blue. Each hemoglobin molecule is made up of four heme groups surrounding a globin group. Heme contains iron and gives a red color to the molecule. Globin consists of two linked pairs of polypeptide chains. The development of each chain is controlled at a separate genetic locus. Changes in the amino acid sequence of these chains results in abnormal hemoglobins. For example, hemoglobin S is found in sickle-cell disease, a severe type of anemia in which the red cells become sickle-shaped when oxygen is in short supply. When red blood cells die, the hemoglobin within them is released and broken up: the iron in hemoglobin is salvaged, transported to the bone marrow by a protein called transferrin and used again in the production of new red blood cells; the remainder of the hemoglobin becomes a chemical called bilirubin that is excreted into the bile which is secreted into the intestine, where it gives the feces their characteristic yellow-brown color.
* * *The red respiratory protein of erythrocytes, consisting of approximately 3.8% heme and 96.2% globin, with a molecular weight of 64,450, which as oxyhemoglobin (HbO2) transports oxygen from the lungs to the tissues where the oxygen is readily released and HbO2 becomes Hb. When Hb is exposed to certain chemicals, its normal respiratory function is blocked; e.g., the oxygen in HbO2 is easily displaced by carbon monoxide, thereby resulting in the formation of fairly stable carboxyhemoglobin (HbCO), as in asphyxiation resulting from inhalation of exhaust fumes from gasoline engines. When the iron in Hb is oxidized from the ferrous to ferric state, as in poisoning with nitrates and certain other chemicals, a nonrespiratory compound, methemoglobin (MetHb), is formed. In humans there are at least five kinds of normal Hb: two embryonic Hb's (Hb Gower-1, Hb Gower-2), fetal (Hb F), and two adult types (Hb A, Hb A2). There are two α globin chains containing 141 amino acid residues, and two of another kind (β, γ, δ, ε, or ζ), each containing 146 amino acid residues in four of the Hb's. Hb Gower-1 has two ζ chains and two ε chains. The production of each kind of globin chain is controlled by a structural gene of similar Greek letter designation; normal individuals are homozygous for the normal allele at each locus. Substitution of one amino acid for another in the polypeptide chain can occur at any codon in any of the five loci and have resulted in the production of many hundreds of abnormal Hb types, most of no known clinical significance. In addition, deletions of one or more amino acid residues are known, as well as gene rearrangements due to unequal crossing over between homologous chromosomes. The Hb types below are the main abnormal types known to be of clinical significance. Newly discovered abnormal Hb types are first assigned a name, usually the location where discovered, and a molecular formula is added when determined. The formula consists of Greek letters to designate the basic chains, with subscript 2 if there are two identical chains; a superscript letter (A if normal for adult Hb, etc.) is added, or the superscript may designate the site of amino acid substitution (numbering amino acid residues from the N-terminus of the polypeptide) and specifying the change, using standard abbreviations for the amino acid s. There is an exhaustive listing of variant hemoglobins in MIM where a composite numbering system is used.- h. A2 [MIM*141850] the normal Hb (Hb A2) of the formula α2Aδ2 or α2δ2, which makes up approximately 2.5% of the total adult h. concentration. At least 18 mutant variants of the δ chain have been reported.- h. anti- Lepore a group of abnormal hemoglobins similar to h. Lepore. These hemoglobins have normal α chains, but the non-α chain consists of the N-terminal portion of the β chain joined to the C-terminal portion of the δ chain. This is the opposite crossing over pattern observed in h. Lepore. Examples of h. anti-Lepore include HbMiyada, Hb PCongo, Hb PNilotic, and HbLincoln Park. There is also one variant that is both h. Lepore and h. anti-Lepore (HbParchman). Cf.:h. Lepore.- h. Bart [MIM*142309] a Hb homotetramer (all four polypeptides identical) of formula γ4, found in the early embryo and in α-thalassemia 2; not effective in oxygen transport; does not display a Bohr effect.- h. C [MIM*141900.0038] an abnormal Hb with substitution of lysyl residue for glutamyl at the 6th position of the β chain, of formula α22Aβ26Glu→Lys, this type reduces the normal plasticity of erythrocytes. Heterozygotes: Hb C trait, about 28–44% of total Hb is Hb C, no anemia. Homozygotes: nearly all Hb is Hb C, moderate normocytic hemolytic anemia. Individuals heterozygous for both Hb C and Hb S (Hb SC disease) and for Hb C and thalassemia are known, and have atypical hemolytic anemias; sickling is enhanced in Hb SC disease.- h. CGeorgetown, h. CHarlem [MIM*141900.0039] two abnormal Hb's, both with the substitution of a valyl residue for a glutamyl residue at the 6th position of the β chain as in Hb S, and in addition, each has a second substitution of an asparaginyl residue for an aspartyl residue at position 73 of the β chain; both types cause sickling of erythrocytes similar to Hb S.- h. Chesapeake (HbChesapeake) [MIM*141800.0018] an abnormal h. with a single α chain substitution, molecular formula α292Arg→Leuβ2A; heterozygotes have polycythemia, apparently to compensate for the increased oxygen affinity of this Hb, resulting in decreased liberation of oxygen in the tissues.- h. Constant Spring an abnormal h. having an extended polypeptide chain (31 additional amino acyl residues) on the α chain (thus, the α chain is 172 amino acid s long); approximately 20% of the individuals with Hb H disease also have this defect.- h. DPunjab [MIM*141900.0065] an abnormal Hb with a single β chain substitution, molecular formula α2Aβ2121Glu→Gln; heterozygotes are asymptomatic, homozygotes have mild hemolytic anemia; there is an increase in O2 affinity; identical to h. DLos Angeles, h. DNorth Carolina, h. DPortugal, h. DChicago, and hemoblogin Oak Ridge.- h. E [MIM*141900.0071] an abnormal Hb with a single β chain substitution, molecular formula α2Aβ226Glu→Lys, common in Southeast Asia, especially Thailand; heterozygotes are asymptomatic with 35–45% Hb E; homozygotes have mild to moderate hemolytic anemia with 90–100% Hb E and the remainder Hb F.- embryonic h. h. Gower-1, h. Gower-2.- h. F [MIM*142200] normal fetal Hb (Hb F) of molecular formula α2Aγ2F, which is the major Hb component during intrauterine life, decreasing rapidly during infancy to reach a concentration of less than 0.5% in normal children and adults; the concentration of Hb F is increased in some hemoglobinopathies and in some cases of hypoplastic anemia, pernicious anemia, and leukemia; Hb F has a weaker affinity for 2,3-bisphosphoglycerate than does Hb A. More than 50 mutant variants of the γ chain have been reported. SYN: fetal h..- h. F (hereditary persistence of) [MIM*142200.0026] a condition due to an allele that depresses synthesis of β and δ chains (as in thalassemia), but this is fully compensated by increased γ chain synthesis and there is no anemia; there are 3 types: 1) African type, no β or δ chain synthesis by the chromosome with the abnormal gene, heterozygotes have 20–30% Hb F and Hb A2 slightly decreased, homozygotes form no Hb A or Hb A2; 2) Greek type, reduced β and δ chain synthesis, heterozygotes have 10–20% Hb F and normal Hb A2; 3) Swiss type, heterozygotes have only 1 to 3% Hb F and normal Hb A2.- glycosylated h. any one of four h. A fractions (AIa1, AIa2, AIb, or AIc) to which d-glucose and related monosaccharides are covalently linked; concentrations are increased in the erythrocytes of patients with diabetes mellitus and can be used as a retrospective index of glucose control over time in such patients.- h. Gower-1 a Hb of molecular formula ζ2ε2, found as a minor Hb in the early embryo; disappears by the third month of pregnancy in favor of h. Gower-2 and hemaglobin Portland and then by Hb F; the ζ chain has 141 amino acid residues. Synthesis of the ζ chain is deficient in cases of hydrops fetalis. Cf.:h. Gower-2, h. Portland.- h. Gower-2 a normal Hb of molecular formula α2Aε2, which is a major Hb component of the early embryo; production of ε chains normally ceases at about the third month of fetal development and is replaced by Hb F. Cf.:h. Gower-1, h. Portland.- green h. SYN: choleglobin.- h. H [MIM*142309] a homotetramer of Hb (all four polypeptides identical) of molecular formula β4, found only when α chain synthesis is depressed and not effective in oxygen transport. Hb H disease (α-thalassemia intermedia) is a thalassemialike syndrome in individuals heterozygous for both severe and mild genes for α-thalassemia; moderate anemia and red cell abnormalities with 25–35% Hb Bart at birth, but with Hb Bart later replaced by Hb H and with Hb A2 decreased. Hb H shows no cooperativity with O2 binding and does not exhibit a Bohr effect.- h. I [MIM*141800.0055] an abnormal Hb with a single α chain substitution, molecular formula α216Lys→Gluβ2A; a thalassemialike syndrome has been found in persons heterozygous for both Hb I and α-thalassemia genes, with formation of about 70% Hb I.- h. JCapetown [MIM*141800.0063] an abnormal Hb with a single α chain substitution, molecular formula α292Arg→Glnβ2A; heterozygotes have polycythemia because of increased oxygen affinity of this Hb.- h. Kansas [MIM*141900.0145] an abnormal Hb of molecular formula α2Aβ2102Asn→Thr; found in association with familial cyanosis due to decreased oxygen affinity of this Hb.- h. Lepore [MIM 142000-various] a group of abnormal Hb's with normal α chains, but the non-α chains consist of the N-terminal portion of the δ chain joined to the C-terminal portion of the β chain, apparently as the result of nonhomologous pairing and crossing over between the genes for β and δ chains. The major types are Hb LeporeBoston (identical to Hb LeporeWashington), Hb LeporeHollandia, and Hb LeporeBaltimore, which differ in the region of crossing over (δ87–β116, δ22–β50, and δ50–β86, respectively). Heterozygotes form about 10% Hb Lepore, normal amounts of Hb A2, and moderately increased amounts of Hb F and usually have mild anemia, microcytosis, and hypochromia; homozygotes form only Hb Lepore and Hb F and have severe anemia. Cf.:h. anti- Lepore.- h. M [MIM*142310 & various] a group of abnormal Hb's in which a single amino acid substitution favors the formation of methemoglobin in spite of normal quantities of methemoglobin reductase. Strictly speaking, Hb's M are hemoglobins with mutations at the proximal or distal histidyl residues. Other Hb's M tend to favor the Fe(III) state. Heterozygotes have congenital methemoglobinemia; the homozygous state of these genes is unknown and is presumably lethal. Specific types include: Hb MIwate, α87His→Tyr (α chain, position 87, histidine replaced by tyrosine); Hb M Hyde Park, β92His→Tyr; Hb MBoston, α58His→Tyr; Hb MSaskatoon, β63His→Tyr; Hb MMilwaukee-1, β67Val→Glu.- mean corpuscular h. (MCH) the h. content of the average red cell, calculated from the h. therein and the red cell count, in erythrocyte indices.- oxygenated h. SYN: oxyhemoglobin.- h. Portland a form of embryonic h. containing the ζ chains of h. Gower-1 and the γ chains of Hb F, thus having the formula ζ2γ2; essentially disappears by the third month of pregnancy. Cf.:h. Gower-1, h. Gower-2.- h. Rainier [MIM*141900-0232] an abnormal Hb of the molecular formula α2Aβ2145Tyr→Cys; heterozygotes have polycythemia because of increased oxygen affinity of this Hb.- reduced h. the form of Hb in red blood cells after the oxygen of oxyhemoglobin is released in the tissues.- h. S [MIM*141900] an abnormal Hb with substitution of valine for glutamic acid at the 6th position of the β chain; the formula is α2Aβ2S, or, more specifically, α2Aβ26Glu→Val. Heterozygous state : sickle cell trait, no anemia, Hb S 20–45% of total, the rest Hb A. Homozygous state : sickle cell anemia, Hb S 75–100% of total, the rest Hb F or Hb A2. SYN: sickle cell h..- unstable hemoglobins a group of rare Hb's with amino acid substitutions (or amino acid deletions in three types) that alter the three-dimensional shape of the globin in a manner that renders the molecule unstable; they have an increased but variable tendency to autooxidation and Heinz body formation and are associated with congenital nonspherocytic hemolytic anemia. The unstable β-chain abnormalities include Hb's Genova, Gun Hill, Hammersmith, Köln, Philly, Sabine, Santa Ana, Sydney, Wien, and Zürich; unstable α-chain abnormalities include Hb's Bibba, Sinai, and Torino.- h. Yakima [MIM*141900-0301] an abnormal Hb of the molecular formula α2Aβ299Asp→His; heterozygotes have polycythemia because of increased oxygen affinity of this Hb.
* * *
he·mo·glo·bin or chiefly Brit hae·mo·glo·bin 'hē-mə-.glō-bən n1) an iron-containing respiratory pigment of vertebrate red blood cells that functions primarily in the transport of oxygen from the lungs to the tissues of the body, that consists of four polypeptide chains of which two are of the type designated alpha and two are of one of the types designated beta, gamma, or delta and each of which is linked to a heme molecule, that combines loosely and reversibly with oxygen in the lungs or gills to form oxyhemoglobin and with carbon dioxide in the tissues to form carbhemoglobin, that in humans is present normally in blood to the extent of 14 to 16 grams in 100 milliliters expressed sometimes on a scale of 0 to 100 with an average normal value (as 15 grams) taken as 100, and that is determined in blood either colorimetrically or by quantitative estimation of the iron present see FETAL HEMOGLOBIN, hemoglobin A compare CARBOXYHEMOGLOBIN, METHEMOGLOBIN2) any of numerous iron-containing respiratory pigments of various organisms (as invertebrates and yeasts)he·mo·glo·bin·ic or chiefly Brit hae·mo·glo·bin·ic .hē-mə-glō-'bin-ik adjhe·mo·glo·bi·nous or chiefly Brit hae·mo·glo·bi·nous -'glō-bə-nəs adj* * *
he·mo·glo·bin (Hb) (heґmo-glo″bin) the red oxygen-carrying pigment of erythrocytes, formed by developing erythrocytes in bone marrow. It is a type of hemoprotein that contains four heme groups and globin and has the property of reversible oxygenation. A molecule of hemoglobin contains four polypeptide globin chains, composed of between 141 and 146 amino acids; those most often found are α and β chains, with γ and δ chains seen somewhat less often. Different types of hemoglobins are determined by different combinations of chains, with the number of chains of each type in the molecule being indicated by a subscript. For example, hemoglobin F (fetal h.), the predominant type in the newborn, may be written α2Aγ2F, and hemoglobin A (adult h.), which is normally predominant in the adult, may be written α2Aβ2A or α2β2. Another hemoglobin, hemoglobin A2> (designated α2Aδ2A2 or α2Aδ2), is usually present in limited minor concentrations. Hundreds of hemoglobins with differing electrophoretic mobilities and characteristics have been reported; the first ones were given capital letters, such as S, C, D, E, G, H, I, J, K, L, M, N, and Q. As refined biochemical techniques led to the discovery of many additional hemoglobins, newer standards for nomenclature were devised: those with electrophoretic mobility equal to one of the lettered hemoglobins could be named with that letter using the place of discovery as a subscript, such as hemoglobin MSaskatoon or hemoglobin MMilwaukee. New hemoglobins with unique electrophoretic mobilities are now often named simply for the laboratory, hospital, or town where they were discovered, such as hemoglobin Chesapeake or hemoglobin Gun Hill. When known, the number of each amino acid substituting in each polypeptide in the molecule should be indicated by the appropriate superscript numeral. Symbol Hb.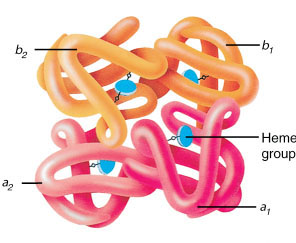
 Hemoglobin, comprising four globin chains, each with a heme group.
Hemoglobin, comprising four globin chains, each with a heme group.
Medical dictionary. 2011.